بیماری نقص انتقال دهنده کارنیتین
Carnitin Transporter Deficiency
دکتر عبدالرضا وارسته، فهیمه تیموری و اعظم شفائی- بخش بیماریهای متابولیک آزمایشگاه پردیس مشهد
دکتر عبدالرضا وارسته- مركز آموزش عالي علوم پزشكي وارستگان مشهد و مركز تحقيقات ايمونولوژي مشهد
مقدمه:
در رابطه با اکسیداسیون اسیدهای چرب و متابولیسم کارنیتین سه بیماری شناسایی گردیده که شامل نقص انتقالدهنده کارنیتین (CTD)، نقص کارنیتین پالمیتیل ترانسفراز یک (CPT1) و نقص کارنیتین پالمیتیل ترانسفراز دو (CPT2) میباشند. این بیماریها در ارتباط با انتقال آسیل کارنیتینها بین سیتوزول و میتوکندری بوجود میآیند. افراد مبتلا به این بیماری برای هضم چربیها و تبدیل آنها به انرژی با مشکل مواجه هستند.

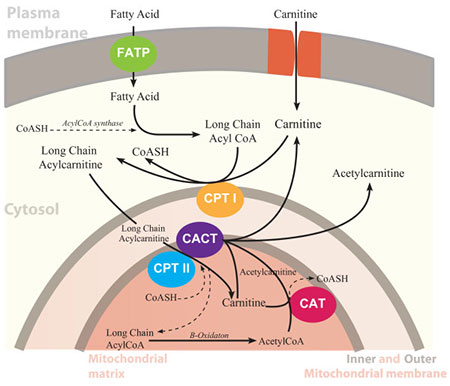
در این مقاله به بررسی بیماری نقص انتقالدهنده کارنیتین یا CTD ((Carnitin Transporter Deficiency میپردازیم. همانطور که اشاره شد، این بیماری اختلالی مرتبط با اکسیداسیون اسیدهای چرب میباشد. از اکسیداسیون اسیدهای چرب در میتوکندری ATP تولید میشود. همچنین این اکسیداسیون آسیلکارنیتینکوآ مورد نیاز گلوکونئوژنز را تأمین میکند. در این بیماری، هضم چربیها و تبدیل آنها به انرژی در مبتلایان به این بیماری بهدرستی صورت نمیگیرد. بیماری CTD در اثر نقــص در آنزیم انتقالدهنــــده کارنیتین (Carnitine-acylcarnitin Translocase )بوجود میآید. برای انتقال آسیلکارنیتین با زنجیره بلند از سیتوپلاسم سلول به ماتریکس میتوکندری (محل بتا اکسیداسیون اسیدهای چرب) حضور این آنزیم انتقالدهنده ضروری است.
نقص در این پروتئین انتقالدهنده، باعث نقص در اکسیداسیون اسیدهای چرب با زنجیره بلند شده و در نتیجه باعث تجمع آسیلکارنیتین با زنجیره بلند در خارج میتوکندری و پلاسما میگردد. اسیدهای چرب با زنجیره کوتاه و متوسط (C8 و کمتر) برای ورود به میتوکندری، نیاز به انتقالدهنده کارنیتین ندارند، بنابراین با وجود این نقص، بدن میتواند از آنها برای تأمین انرژی استفاده کند. کارنیتین منتقل شده به هضم چربیهای موجود در مواد غذایی و تبدیل آنها به انرژی کمک میکند. همچنین کارنیتین در تجزیه چربیهای ذخیرهای بدن و تبدیل آنها به انرژی نقش دارد.
چربیها، منبع اصلی تولید انرژی در هنگام گرسنگی و خواب هستند. هنگامی که آنزیم انتقالدهنده کارنیتین بهدرستی عمل نکند و یا اصلاً وجود نداشته باشد، تنها منبع تولید انرژی در بدن گلوکز خواهد بود. گلوکز یک منبع خوب انرژی است، اما مقدار آن در بدن محدود است. هنگامی که سطح گلوکز خون (منبع اصلی انرژی بدن) افت کند، بدن برای تولید انرژی، از چربیها استفاده میکند، بنابراین بیماری CTD باعث بروز علائمی در مبتلایان به این بیماری میشود.
علائم بالینی:
بیماریCTD دو فرم بالینی مختلف دارد. نوع اول در دوران نوزادی و نوع دوم در دوران کودکی بروز میکند.
بیماری CTD در نوزادان:
زمان بروز اولین علائم در این فرم بیماری، معمولاً از بدو تولد تا 3 سالگی است. در بیماران مبتلا به CTD، حملات ناگهانی و پیشبینینشده بیماری دیده میشود. این حملات ناگهانی، بحرانهای متابولیکی نامیده میشوند. برخی از علائم بحرانهای متابولیکی شامل خوابآلودگی مفرط، کاهش اشتها، بیقراری، تحریکپذیری و تغییرات رفتاری میشوند. علائم دیگری که در ادامه ایجاد میشوند شامل تب، حالت تهوع، اسهال، استفراغ و هیپوگلیسمی هستند. اگر بحرانهای متابولیکی درمان نشوند، ممکن است باعث بروز مشکلاتی همچون مشکلات تنفسی، تورم مغز، تشنج و یا کوما شوند. کودکانی که تحتدرمان قرار نگرفته اند ممکن است علائمی مانند بزرگی قلب، بزرگی کبد، ضعف ماهیچهها و کمخونی را بروز دهند. تکرار بحرانهای متابولیکی میتواند باعث آسیبهای دائمی مغزی و در نتیجه ناتوانی در یادگیری و یا عقبماندگی ذهنی این کودکان شود. علائم بحرانهای متابولیکی، معمولاً بعد از یک دوره گرسنگی و یا در هنگام عفونت و بیماری بروز میکنند.
بیماری CTD در کودکان:
کودکان مبتلا به این فرم از بیماری، تا زمان بروز علائم کاملاً سالم به نظر میرسند. زمان بروز اولین علائم بیماری، معمولاً بین 1 تا 7 سالگی است. این علائم معمولاً شامل بزرگی کبد و ضعف ماهیچهها میشوند. اگر این بیماری درمان نشود، ممکن است باعث نارسایی قلبی و حتی مرگ این کودکان شود. بیمارانــــــــی که به CTD نوع کودکان مبتلا هستند، دچار افت قند خون و بحرانهای متابولیکی نمیشوند. همچنین این فرم از بیماری، هوش بیماران مبتلا را تحت تأثیر قرار نمیدهد. در تعدادی از بیماران، هیچگاه علائم بیماری بروز نمیکند و این افراد معمولاً به دنبال شناسایی یکی از اعضای خانواده آنها که مبتلا به این بیماری است، شناسایی میشوند.
روش تشخیص:
روش نوین غربالگری نوزادان با استفاده از تکنیک TMS* یا MS/MS، افزایشی در سطح چند نوع از آسیل کارنیتینهای با زنجیره بلند (مثل C16، C18،C18:1 وC18:2) را نشان میدهد. این یافتهها مشخصهCTD هستند اما قطعی نیستند، چرا که درCPT-2Type2)Carnitine Palmitoyl Transferase) نیز یافتههای مشابهی دیده میشود. تشخیص قطعی بیماری CTD نیاز به کشت فیبروبلاستها یا بررسی موتاسیونهای DNA دارد. تشخیص قبل از تولد بوسیله شناسایی موتاسیونها در والدین امکان پذیر است.
درمان:
هدف کلی درمان در این بیماران، پیشگیری از هیپوگلیسمی و محدودیت استفاده از اسیدهای چرب با زنجیره بلند میباشد. در این خصوص توجه به موارد زیر کمک کننده است:
- مصرف L-Carnitine: این دارو یک ماده طبیعی مفید است که به تولید انرژی در بافتها و رهایی بدن از مواد مضر کمک میکند. این دارو میتواند مشکلات قلبی و ضعف ماهیچههای بیماران مبتلا به CTD را کاهش دهد.
- پرهیز از گرسنگی طولانی مدت کودک: کودکان مبتلا به CTD باید به طور متناوب غذا بخورند تا از بحران متابولیکی در آنها جلوگیری شود. به طور کلی اغلب کودکان مبتلا باید هر 6-4 ساعت یک بار غذا بخورند. مبتلایان حتی در طول شب نیز نباید گرسنه بمانند. معمولاً نوجوانان و بزرگسالان مبتلا به CTD در صورتی که دیگر مراحل درمان را به دقت پیگیری نمایند، میتوانند تا 12 ساعت گرسنگی را بدون مشکل تحمل کنند.
رژیم غذایی:
گاهی یک رژیم غذایی حاوی کربوهیدرات زیاد و چربی کم، به این بیماران توصیه میشود. هرگونه تغییر در رژیم غذایی این بیماران، باید با اجازه متخصص تغذیه باشد.
کودکان مبتلا به CTD در هنگام بیماری، حتی اگر گرسنه نباشند، باید مقادیر بیشتری مایعات و غذاهای نشاستهای مصرف کنند، در غیر اینصورت ممکن است دچار افت قند خون و بحرانهای متابولیکی شوند. بسیاری از کودکان در دوران بیماری تمایلی به خوردن غذا ندارند، در این صورت باید فوراً آنها را به بیمارستان منتقل نمود.
بیماران مبتلا به CTD در صورتی که درمان فوری و مداوم داشته باشند، اغلب زندگی عادی همراه با رشد و نمو طبیعی خواهند داشت. مصرف L-Carnitine بسیاری از مشکلات ماهیچهای و قلبی این بیماران را کاهش خواهد داد. در تعدادی از بیماران تحت درمان، ممکن است باز هم بحرانهای متابولیکی دیده شوند. تکرار این حملات، میتواند باعث ایجاد مشکلات دائمی یادگیری و یا عقبماندگی ذهنی در این افراد شود.
توارث:
اين نقص معمولاً از الگوي وراثتي اتوزومال مغلوب پيروي ميكند. در اين الگو بيماران مبتلا باید دو كپي از ژن جهش يافته را براي بروز علائم داشته باشند. افرادي كه داراي يك ژن معیوب هستند، ناقل خوانده ميشوند و علائم بیماری را نشان نميدهند، اما ميتوانند ژن را به فرزندان خود منتقل كنند. وقتی هر دو والدین ناقل باشند، 25% احتمال تولد کودک سالم، 50% احتمال تولد کودک ناقل و 25% احتمال تولد کودک مبتلا به بیماری CTD، در هر بارداری وجود دارد.

الگوی وراثتی اتوزوم مغلوب
انجام آزمایش در طول بارداری:
اگر یکی از فرزندان خانواده مبتلا به CTD باشد، میتوان آزمایش ژنتیک را در طول بارداری بعدی انجام داد. نمونه مورد نیاز برای انجام این تست از آمینوسنتز و یا CVS به دست میآید.
زنان بارداری که جنین آنها مبتلا به CTD است، ممکن است علائمی همچون: استفراغ بیش از حد، درد در ناحیه شکم، افزایش فشار خون، زردی، خونریزی شدید و ذخیرهسازی غیرطبیعی چربی در کبد (توسط بیوپسی مشخص میشود) را داشته باشند.
شیوع بیماری:
برخی منابع شیوع این بیماری را حدود 1 مورد در هر40,000 تولد ذکر کردهاند، اما پیش بینی میشود شیوع این بیماری در کشورهایی همانند ایران، که ازدواجهای فامیلی رواج بیشتری دارند، بیشتر باشد.
طبق بررسیهای انجام شده در آزمایشگاه پردیس مشهد از بین 1172 کودک که توسط تکنیک TMS غربالگری شده بودند، 5 نفر مبتلا به بیماری CTD شناسایی شدند. بدیهی است که این تعداد بیمار و روش بررسی آنها، نمیتواند میزان شیوع این بیماری را در جامعه ما مشخص نماید، اما میتواند بيانگر شیوع بالاتر این نقص، در جامعه ما نسبت به شیوع جهانی آن باشد. یقیناً استفاده از روشهای غربالگری پیشرفته میتواند تصویر دقیق تری از شیوع این بیماری در جامعه ما ارائه نماید.
با توجه به اهمیت زمان، در درمان و پیشگیری از عوارض سوء این بیماری و تحمیل هزینههای روحی و مالی این بیماری به خانواده و جامعه، منطقی به نظر میرسد که غربالگری این بیماری همانند اکثر کشورهای پیشرفته دنیا و اغلب کشورهای همسایه، در برنامه غربالگریهای روتین برای تمام نوزادان متولد شده در ایران قرار گیرد.
Tandem Mass Spectrometry *
: References
1) Perkin Elmer Genetics, Newborn Screening
2) American College of Medical Genetics, 2006
3) Fenton, W.A., Gravel, R.A. and Rosenblatt, D.S. Disorder of Propionate and Methylmalonate Metabolism. In, The Metabolic and Molecular basis of inherited Disease.
4) Kahler, S.G., Millington, D.S., Cederbaum, S.D., et al. Parenteral nutrition in propionic and methylmalonic academia.
5) Keletzko, B., Bachmann, C. and Wendel, U. Antibiotic therapy for improvement of metabolic control in methylmalonic aciduria.
6) Roe, C.R., Hoppel, C.L., Stacey, T.E., et al. Metabolic response to carnitine in methylmalonic aciduria.
7) Sniderman, L.C., Lambert, M., Giguere, R., et al. Outcome of individuals with low-moderate methylmalonic aciduria detected through a neonatal screening program.
